You signed in with another tab or window. Reload to refresh your session.You signed out in another tab or window. Reload to refresh your session.You switched accounts on another tab or window. Reload to refresh your session.Dismiss alert
170
+
169
171
### Electrostatic Interaction
172
+
170
173
Interaction between charged particles obeys Coulomb's law. The movement of such bodies can be simulated using `ChargedParticle` and `ChargedParticles` structures.
171
174
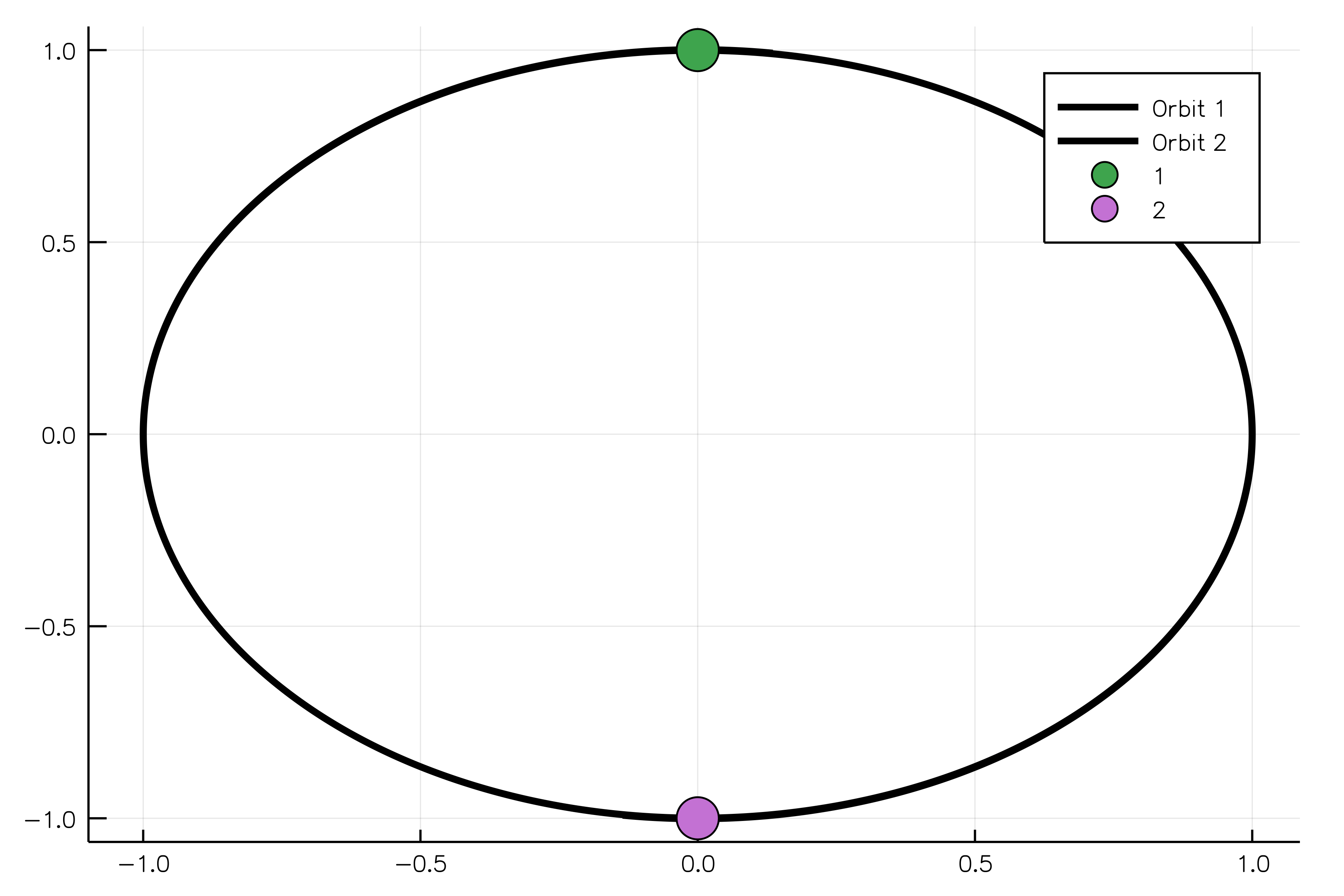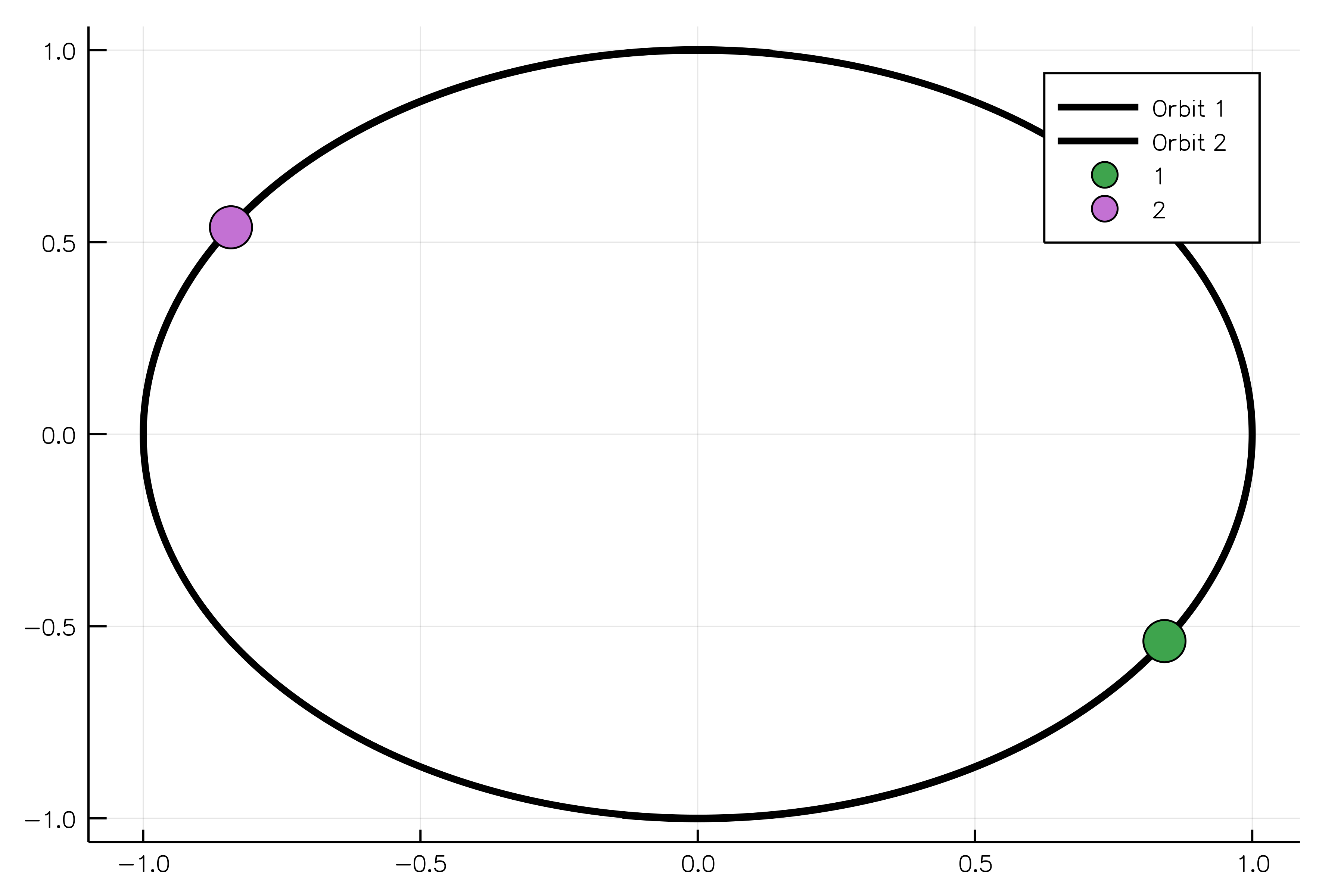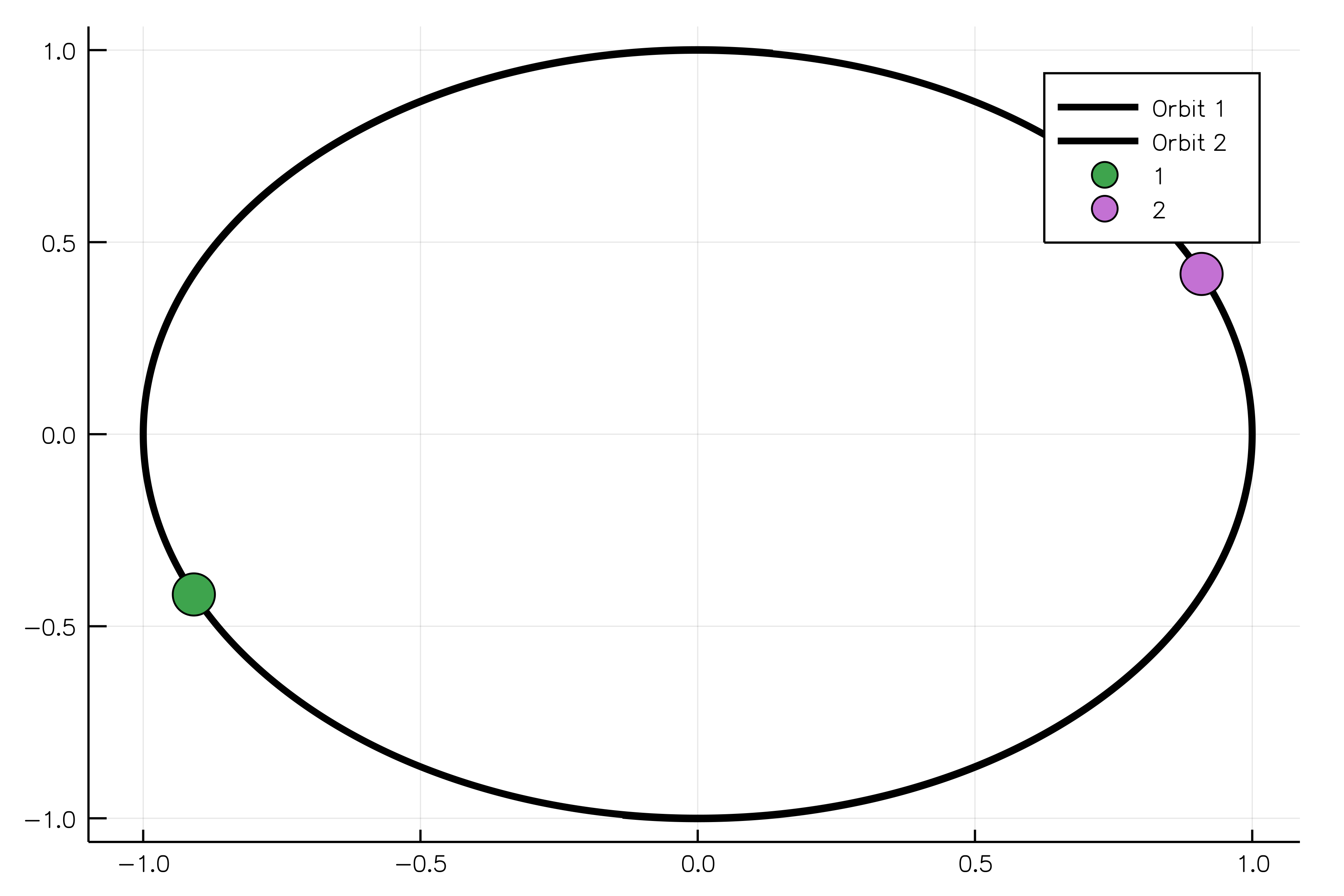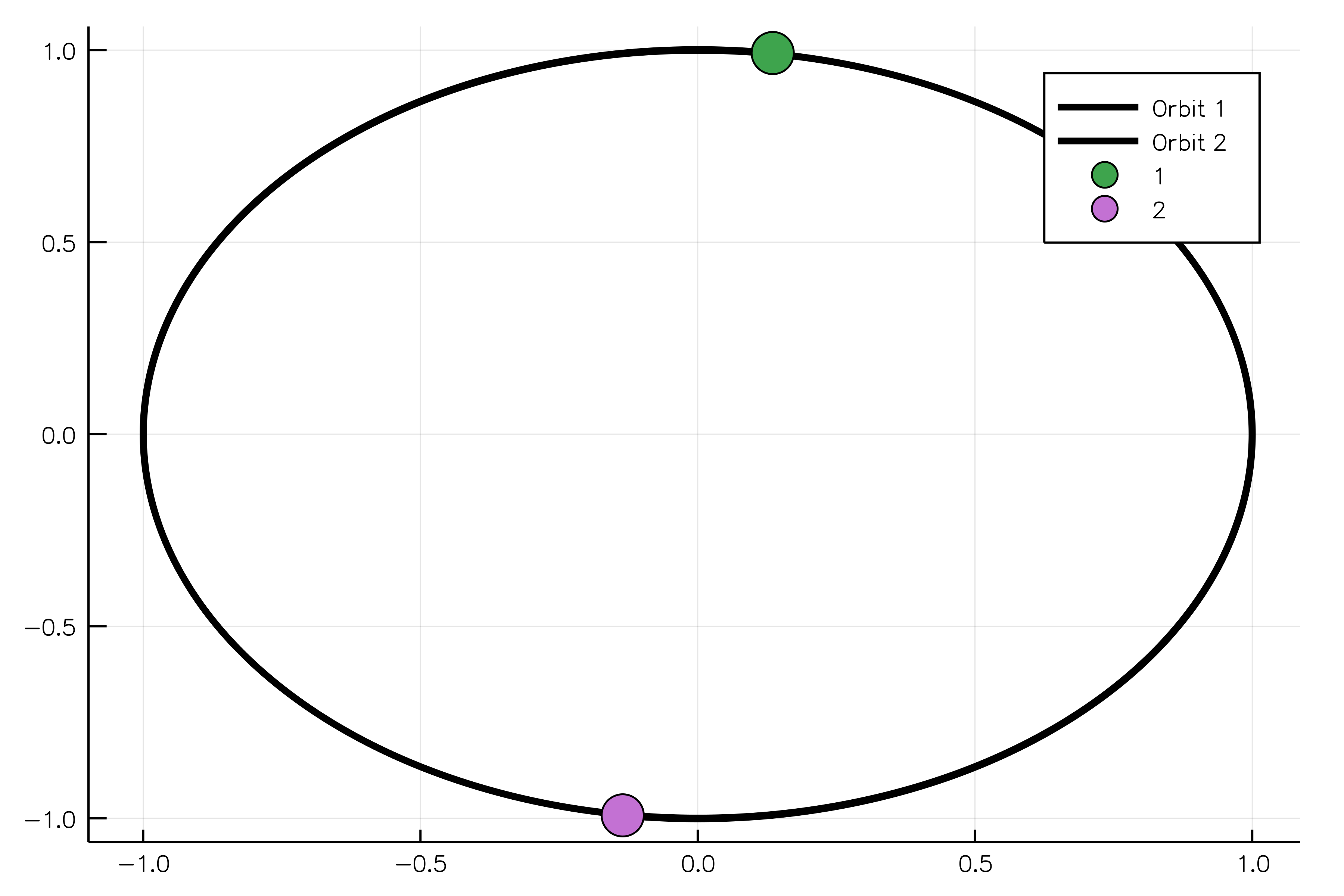
The following example shows how to model two oppositely charged particles. If one body is more massive than another, it will be possible to observe the rotation of the light body around the heavy one without adjusting their positions in space. The constructor for the `ChargedParticles` system requires bodies and Coulomb's constant `k` to be passed as arguments.
175
+
172
176
```julia
173
177
r =100.0# m
174
178
q1 =1e-3# C
@@ -183,9 +187,12 @@ system = ChargedParticles([p1, p2], k)
183
187
simulation =NBodySimulation(system, (0.0, t))
184
188
sim_result =run_simulation(simulation)
185
189
```
190
+
186
191
### Magnetic Interaction
192
+
187
193
An N-body system consisting of `MagneticParticle`s can be used for the simulation of interacting magnetic dipoles, though such dipoles cannot rotate in space. Such a model can represent single domain particles interacting under the influence of a strong external magnetic field.
188
194
To create a magnetic particle, one specifies its location in space, velocity, and the vector of its magnetic moment. The following code shows how we can construct an iron particle:
195
+
189
196
```julia
190
197
iron_dencity =7800# kg/m^3
191
198
magnetization_saturation =1.2e6# A/m
@@ -195,27 +202,35 @@ v = SVector(0.0, 0.0, 0.0) # m/s
195
202
magnetic_moment =SVector(0.0, 0.0, magnetization_saturation * mass / iron_dencity) # A*m^2
196
203
p1 =MagneticParticle(r, v, mass, magnetic_moment)
197
204
```
205
+
198
206
For the second particle, we will use a shorter form:
To calculate magnetic interactions properly, one should also specify the value for the constant μ0/4π or its substitute. Having created parameters for the magnetostatic potential, one can now instantiate a system of particles that should interact magnetically. For that purpose, we use `PotentialNBodySystem` and pass particles and potential parameters as arguments.
214
+
204
215
```julia
205
216
parameters =MagnetostaticParameters(μ_4π)
206
217
system =PotentialNBodySystem([p1, p2], Dict(:magnetic=> parameters))
NBodySimulator allows one to conduct molecular dynamic simulations for the Lennard-Jones liquids, the SPC/Fw model of water, and other molecular systems thanks to implementations of basic interaction potentials between atoms and molecules:
225
+
212
226
- Lennard-Jones
213
227
- electrostatic and magnetostatic
214
228
- harmonic bonds
215
229
- harmonic valence angle generated by pairs of bonds
216
-
The comprehensive examples of liquid argon and water simulations can be found in the `examples` folder.
217
-
Here only the basic principles of the molecular dynamics simulations using NBodySimulator are presented using liquid argon as a classical MD system for beginners.
218
-
First, one needs to define the parameters of the simulation:
230
+
The comprehensive examples of liquid argon and water simulations can be found in the `examples` folder.
231
+
Here only the basic principles of the molecular dynamics simulations using NBodySimulator are presented using liquid argon as a classical MD system for beginners.
232
+
First, one needs to define the parameters of the simulation:
233
+
219
234
```julia
220
235
T =120.0# °K
221
236
T0 =90.0# °K
@@ -233,110 +248,156 @@ bodies = generate_bodies_in_cell_nodes(N, m, v_dev, L)
233
248
t1 =0.0
234
249
t2 =2000τ
235
250
```
251
+
236
252
Liquid argon consists of neutral molecules, so the Lennard-Jones potential runs their interaction:
result =run_simulation(simulation, VelocityVerlet(), dt = τ)
247
266
```
267
+
248
268
It is recommended to use `CubicPeriodicBoundaryConditions` since cubic boxes are among the most popular boundary conditions in MD. There are different variants of the `NBodySimulation` constructor for MD:
The default boundary conditions are `InfiniteBox` without any limits, the default thermostat is `NullThermostat` (which does no thermostating), and the default Boltzmann constant `kb` equals its value in SI, i.e., 1.38e-23 J/K.
278
+
256
279
## Water Simulations
280
+
257
281
In NBodySImulator the [SPC/Fw water model](http://www.sklogwiki.org/SklogWiki/index.php/SPC/Fw_model_of_water) is implemented. For using this model, one has to specify parameters of the Lennard-Jones potential between the oxygen atoms of water molecules, parameters of the electrostatic potential for the corresponding interactions between atoms of different molecules, and parameters for harmonic potentials representing bonds between atoms and the valence angle made from bonds between hydrogen atoms and the oxygen atom.
water =WaterSPCFw(bodies, mH, mO, qH, qO, jl_parameters, e_parameters, spc_parameters);
264
289
```
290
+
265
291
For each water molecule here, `rOH` is the equilibrium distance between a hydrogen atom and the oxygen atom, `∠HOH` denotes the equilibrium angle made of those two bonds, `k_bond` and `k_angle` are the elastic coefficients for the corresponding harmonic potentials.
266
292
Further, one can pass the water system into the `NBodySimulation` constructor as a usual system of N-bodies.
Usually, during the simulation, a system is required to be at a particular temperature. NBodySimulator contains several thermostats for that purpose. Here the thermostating of liquid argon is presented, for thermostating of water, one can refer to [this post](https://mikhail-vaganov.github.io/gsoc-2018-blog/2018/08/06/thermostating.html).
Once the simulation is completed, one can analyze the result and obtain some useful characteristics of the system.
300
343
The function `run_simulation` returns a structure containing the initial parameters of the simulation and the solution of the differential equation (DE) required for the description of the corresponding system of particles. There are different functions that help to interpret the solution of DEs into physical quantities.
301
344
One of the main characteristics of a system during molecular dynamics simulations is its thermodynamic temperature. The value of the temperature at a particular time `t` can be obtained by calling this function:
345
+
302
346
```julia
303
347
T =temperature(result, t)
304
348
```
349
+
305
350
### [Radial distribution functions](https://en.wikipedia.org/wiki/Radial_distribution_function)
351
+
306
352
The RDF is another popular and essential characteristic of molecules or similar systems of particles. It shows the reciprocal location of particles averaged by the time of simulation.
353
+
307
354
```julia
308
355
(rs, grf) =rdf(result)
309
356
```
357
+
310
358
The dependence of `grf` on `rs` shows the radial distribution of particles at different distances from an average particle in a system.
311
359
Here, the radial distribution function for the classic system of liquid argon is presented:
312
360
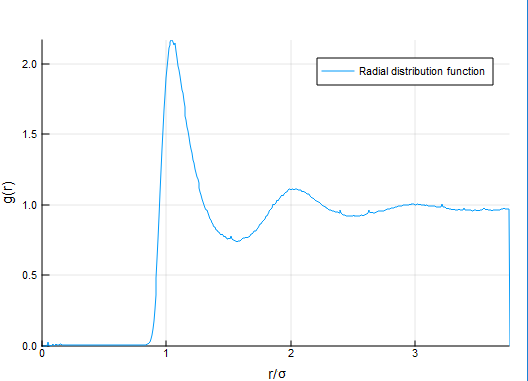
361
+
313
362
### Mean Squared Displacement (MSD)
363
+
314
364
The MSD characteristic can be used to estimate the shift of particles from their initial positions.
365
+
315
366
```julia
316
367
(ts, dr2) =msd(result)
317
368
```
369
+
318
370
For a standard liquid argon system, the displacement grows with time:
319
371
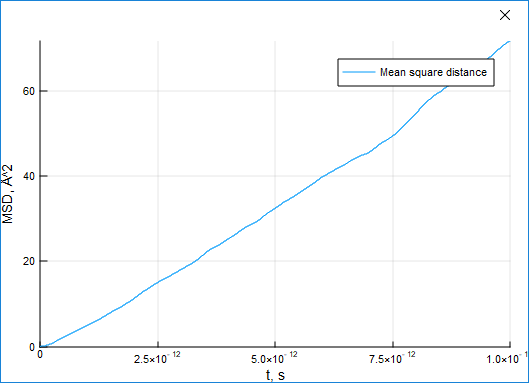
372
+
320
373
### Energy Functions
374
+
321
375
Energy is a highly important physical characteristic of a system. The module provides four functions to obtain it, though the `total_energy` function just sums potential and kinetic energy:
376
+
322
377
```julia
323
378
e_init =initial_energy(simulation)
324
379
e_kin =kinetic_energy(result, t)
325
380
e_pot =potential_energy(result, t)
326
381
e_tot =total_energy(result, t)
327
382
```
383
+
328
384
## Plotting Images
385
+
329
386
Using the tools of NBodySimulator, one can export the results of a simulation into a [Protein Database File](https://en.wikipedia.org/wiki/Protein_Data_Bank_(file_format)). [VMD](http://www.ks.uiuc.edu/Research/vmd/) is a well-known tool for visualizing molecular dynamics, which can read data from PDB files.
387
+
330
388
```julia
331
389
save_to_pdb(result, "path_to_a_new_pdb_file.pdb")
332
390
```
391
+
333
392
In the future, it will be possible to export results via the FileIO interface and its `save` function.
334
393
Using Plots.jl, one can draw the positions of particles at any time of simulation or create an animation of moving particles, molecules of water:
394
+
335
395
```julia
336
396
using Plots
337
397
plot(result)
338
398
animate(result, "path_to_file.gif")
339
399
```
400
+
340
401
Makie.jl also has a recipe for plotting the results of N-body simulations. The [example](https://github.com/MakieOrg/Makie.jl/blob/master/ReferenceTests/src/tests/recipes.jl) is presented in the documentation.
341
402
342
403
## Contributing
@@ -356,40 +417,51 @@ Makie.jl also has a recipe for plotting the results of N-body simulations. The [
356
417
+ See also [SciML Community page](https://sciml.ai/community/)
357
418
358
419
## Reproducibility
420
+
359
421
```@raw html
360
422
<details><summary>The documentation of this SciML package was built using these direct dependencies,</summary>
361
423
```
424
+
362
425
```@example
363
426
using Pkg # hide
364
427
Pkg.status() # hide
365
428
```
429
+
366
430
```@raw html
367
431
</details>
368
432
```
433
+
369
434
```@raw html
370
435
<details><summary>and using this machine and Julia version.</summary>
371
436
```
437
+
372
438
```@example
373
439
using InteractiveUtils # hide
374
440
versioninfo() # hide
375
441
```
442
+
376
443
```@raw html
377
444
</details>
378
445
```
446
+
379
447
```@raw html
380
448
<details><summary>A more complete overview of all dependencies and their versions is also provided.</summary>
0 commit comments